
Ao editor - Desde os primeiros relatos de uma nova pneumonia (COVID-19) em Wuhan, província de Hubei, China 1 , 2 , houve uma discussão considerável sobre a origem do vírus causador, SARS-CoV-2 3 (também conhecido como como HCoV-19) 4 . Atualmente, as infecções por SARS-CoV-2 são generalizadas e, em 11 de março de 2020, 121.564 casos foram confirmados em mais de 110 países, com 4.373 mortes 5 .
SARS-CoV-2 é o sétimo coronavírus conhecido por infectar seres humanos; SARS-CoV, MERS-CoV e SARS-CoV-2 podem causar doença grave, enquanto HKU1, NL63, OC43 e 229E estão associados a sintomas leves 6 . Aqui, revisamos o que pode ser deduzido sobre a origem do SARS-CoV-2 a partir da análise comparativa de dados genômicos. Oferecemos uma perspectiva sobre os recursos notáveis do genoma SARS-CoV-2 e discutimos cenários pelos quais eles poderiam ter surgido. Nossas análises mostram claramente que o SARS-CoV-2 não é uma construção de laboratório ou um vírus propositadamente manipulado.
Características notáveis do genoma SARS-CoV-2
Nossa comparação de alfa e betacoronavírus identifica duas características genômicas notáveis do SARS-CoV-2: (i) com base nos estudos estruturais 7 , 8 , 9 e nos experimentos bioquímicos 1 , 9 , 10 , o SARS-CoV-2 parece ser otimizado para ligação ao receptor humano ACE2; e (ii) a proteína spike de SARS-CoV-2 possui um local de clivagem polibásico (furina) funcional no limite S1 – S2 através da inserção de 12 nucleotídeos 8 , o que adicionalmente levou à aquisição prevista de três glicanos ligados ao O o site.
1. Mutações no domínio de ligação ao receptor da SARS-CoV-2
O domínio de ligação ao receptor (RBD) na proteína spike é a parte mais variável do genoma do coronavírus 1 , 2 . Demonstrou-se que seis aminoácidos RBD são críticos para a ligação a receptores ACE2 e para determinar a faixa hospedeira de vírus do tipo SARS-CoV 7 . Com coordenadas baseadas em SARS-CoV, elas são Y442, L472, N479, D480, T487 e Y4911, que correspondem a L455, F486, Q493, S494, N501 e Y505 em SARS-CoV-2 7 . Cinco destes seis resíduos diferem entre SARS-CoV-2 e SARS-CoV (Fig. 1a ). Com base nos estudos estruturais 7 , 8 , 9 e nos experimentos bioquímicos 1 , 9 , 10, O SARS-CoV-2 parece ter uma RBD que se liga com alta afinidade à ACE2 de humanos, furões, gatos e outras espécies com alta homologia de receptores 7 .
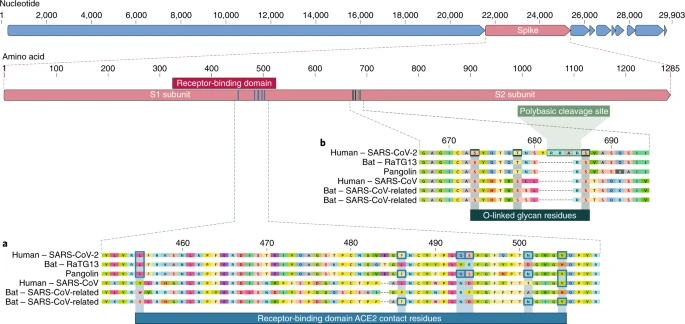
a , Mutações nos resíduos de contato da proteína spike SARS-CoV-2. A proteína spike de SARS-CoV-2 (barra vermelha na parte superior) foi alinhada contra os coronavírus mais semelhantes ao SARS-CoV e o próprio SARS-CoV. Os resíduos principais da proteína spike que fazem contato com o receptor ACE2 são marcados com caixas azuis no SARS-CoV-2 e em vírus relacionados, incluindo o SARS-CoV (cepa Urbani). b , aquisição de sítio de clivagem polibásica e glicanos ligados a O. Tanto o local de clivagem polibásica quanto os três glicanos ligados ao O preditos adjacentes são exclusivos do SARS-CoV-2 e não eram vistos anteriormente nos betacoronavírus da linhagem B. As sequências mostradas são do NCBI GenBank, códigos de acesso MN908947 , MN996532 , AY278741 , KY417146e MK211376 . As sequências de coronavírus de pangolim são um consenso gerado a partir de SRR10168377 e SRR10168378 (NCBI BioProject PRJNA573298 ) 29 , 30 .
Enquanto as análises acima sugerem que o SARS-CoV-2 pode se ligar à ACE2 humana com alta afinidade, as análises computacionais prevêem que a interação não é ideal 7 e que a sequência RBD é diferente das mostradas no SARS-CoV como sendo ideal para a ligação ao receptor 7. , 11 . Assim, a ligação de alta afinidade da proteína spike SARS-CoV-2 à ACE2 humana é provavelmente o resultado da seleção natural de uma ACE2 humana ou semelhante a humano que permite que outra solução ótima de ligação surja. Esta é uma forte evidência de que o SARS-CoV-2 não é o produto de manipulação intencional.
2. Local de clivagem da furina polibásica e glicanos ligados ao O
A segunda característica notável do SARS-CoV-2 é um local de clivagem polibásico (RRAR) na junção de S1 e S2, as duas subunidades do espigão 8 (Fig. 1b ). Isso permite uma clivagem eficaz pela furina e outras proteases e tem um papel na determinação da infecciosidade viral e do intervalo de hospedeiros 12 . Além disso, uma prolina líder também é inserida neste site no SARS-CoV-2; assim, a sequência inserida é PRRA (Fig. 1b ). Prevê-se que o turno criado pela prolina resulte na adição de glicanos ligados ao O aos S673, T678 e S686, que flanqueiam o local da clivagem e são exclusivos da SARS-CoV-2 (Fig. 1b) Não foram observados locais de clivagem polibásica nos betacoronavírus relacionados à 'linhagem B', embora outros betacoronavírus humanos, incluindo HKU1 (linhagem A), tenham esses locais e glicanos ligados ao O previstos 13 . Dado o nível de variação genética no pico, é provável que vírus do tipo SARS-CoV-2 com locais de clivagem polibásicos parciais ou completos sejam descobertos em outras espécies.
A conseqüência funcional do local de clivagem polibásica no SARS-CoV-2 é desconhecida e será importante determinar seu impacto na transmissibilidade e patogênese em modelos animais. Experimentos com SARS-CoV mostraram que a inserção de um local de clivagem de furina na junção S1-S2 melhora a fusão célula-célula sem afetar a entrada viral 14 . Além disso, a clivagem eficiente do pico de MERS-CoV permite que os coronavírus do tipo MERS de morcegos infectem células humanas 15 . Nos vírus da gripe aviária, a replicação e transmissão rápidas em populações de frangos altamente densas selecionam a aquisição de locais de clivagem polibásica na proteína hemaglutinina (HA) 16, que tem uma função semelhante à da proteína de pico de coronavírus. A aquisição de locais de clivagem polibásica no AH, por inserção ou recombinação, converte vírus da influenza aviária de baixa patogenicidade em formas altamente patogênicas 16 . A aquisição de locais de clivagem polibásica pelo HA também foi observada após repetidas passagens na cultura celular ou através de animais 17 .
A função dos glicanos ligados ao O previstos não é clara, mas eles podem criar um 'domínio semelhante à mucina' que protege epítopos ou resíduos-chave na proteína spike SARS-CoV-2 18 . Vários vírus utilizam domínios semelhantes à mucina, pois os escudos de glicanos envolvem imuno-invasão 18 . Embora a previsão de glicosilação ligada ao O seja robusta, são necessários estudos experimentais para determinar se esses locais são utilizados no SARS-CoV-2.
Teorias de origens SARS-CoV-2
É improvável que o SARS-CoV-2 tenha surgido através da manipulação laboratorial de um coronavírus semelhante ao SARS-CoV. Como observado acima, o RBD do SARS-CoV-2 é otimizado para ligação ao ACE2 humano com uma solução eficiente diferente das previamente previstas 7 , 11 . Além disso, se a manipulação genética tivesse sido realizada, um dos vários sistemas genéticos reversos disponíveis para os betacoronavírus provavelmente teria sido utilizado 19 . No entanto, os dados genéticos mostram irrefutavelmente que o SARS-CoV-2 não é derivado de nenhum backbone de vírus usado anteriormente 20. Em vez disso, propomos dois cenários que podem explicar de maneira plausível a origem do SARS-CoV-2: (i) seleção natural em um hospedeiro animal antes da transferência zoonótica; e (ii) seleção natural em humanos após transferência zoonótica. Também discutimos se a seleção durante a passagem poderia ter causado o SARS-CoV-2.
1. Seleção natural em um hospedeiro animal antes da transferência zoonótica
Como muitos casos iniciais de COVID-19 foram vinculados ao mercado Huanan em Wuhan 1 , 2 , é possível que uma fonte animal estivesse presente nesse local. Dada a semelhança do SARS-CoV-2 com os coronavírus do tipo SARS-CoV 2 , é provável que os morcegos sirvam como hospedeiros de reservatório para seu progenitor. Embora o RaTG13, amostrado de um bastão de Rhinolophus affinis 1 , seja aproximadamente 96% idêntico ao SARS-CoV-2, seu pico diverge no RBD, o que sugere que ele pode não se ligar eficientemente à ACE2 humana 7 (Fig. 1a ).
Os pangolins malaios ( Manis javanica ) importados ilegalmente na província de Guangdong contêm coronavírus semelhantes ao SARS-CoV-2 21 . Embora o vírus do morcego RaTG13 permaneça o mais próximo do SARS-CoV-2 no genoma 1 , alguns coronavírus de pangolim exibem forte semelhança com o SARS-CoV-2 na RBD, incluindo todos os seis principais resíduos de RBD 21 (Fig. 1 ). Isso mostra claramente que a proteína spike SARS-CoV-2 otimizada para ligação à ACE2 semelhante a humanos é o resultado da seleção natural.
Nem os betacoronavírus de morcego nem os betacoronavírus de pangolim amostrados até agora têm locais de clivagem polibásicos. Embora não tenha sido identificado nenhum coronavírus animal que seja suficientemente semelhante para ter atuado como progenitor direto do SARS-CoV-2, a diversidade de coronavírus em morcegos e outras espécies é muito pouco amostrada. Mutações, inserções e deleções podem ocorrer próximas à junção S1 – S2 dos coronavírus 22, que mostra que o local de clivagem polibásica pode surgir por um processo evolutivo natural. Para que um vírus precursor adquira o local de clivagem polibásica e as mutações na proteína spike adequada para ligação à ACE2 humana, um hospedeiro animal provavelmente teria que ter uma alta densidade populacional (para permitir que a seleção natural procedesse com eficiência) e uma codificação para ACE2 gene que é semelhante ao ortólogo humano.
2. Seleção natural em humanos após transferência zoonótica
É possível que um progenitor de SARS-CoV-2 tenha pulado em seres humanos, adquirindo as características genômicas descritas acima por meio de adaptação durante a transmissão homem-a-homem não detectada. Uma vez adquiridas, essas adaptações permitiriam que a pandemia decolasse e produzisse um conjunto de casos suficientemente grande para acionar o sistema de vigilância que a detectou 1 , 2 .
Todos os genomas de SARS-CoV-2 sequenciados até agora têm as características genômicas descritas acima e, portanto, são derivados de um ancestral comum que os possuía também. A presença em pangolins de uma RBD muito semelhante à da SARS-CoV-2 significa que podemos inferir que isso também ocorreu provavelmente no vírus que pulou para os seres humanos. Isso deixa a inserção do local de clivagem polibásica durante a transmissão de homem para homem.
As estimativas do tempo do ancestral comum mais recente do SARS-CoV-2, feito com os dados atuais da sequência, apontam para o surgimento do vírus no final de novembro de 2019 a início de dezembro de 2019 23 , compatível com os primeiros casos confirmados retrospectivamente 24. Portanto, esse cenário pressupõe um período de transmissão não reconhecida em humanos entre o evento zoonótico inicial e a aquisição do local de clivagem polibásica. Oportunidades suficientes poderiam ter surgido se houvesse muitos eventos zoonóticos anteriores que produzissem cadeias curtas de transmissão de homem para homem por um período prolongado. Essa é essencialmente a situação do MERS-CoV, para o qual todos os casos humanos são o resultado de repetidos saltos do vírus de camelos dromedários, produzindo infecções únicas ou cadeias de transmissão curtas que acabam resolvendo, sem adaptação à transmissão sustentada 25 .
Estudos de amostras humanas depositadas podem fornecer informações sobre se essa propagação enigmática ocorreu. Estudos sorológicos retrospectivos também podem ser informativos, e alguns estudos foram realizados mostrando exposições de baixo nível a coronavírus do tipo SARS-CoV em certas áreas da China 26 . Criticamente, no entanto, esses estudos não poderiam ter distinguido se as exposições eram devidas a infecções anteriores com SARS-CoV, SARS-CoV-2 ou outros coronavírus do tipo SARS-CoV. Estudos sorológicos adicionais devem ser conduzidos para determinar a extensão da exposição humana prévia à SARS-CoV-2.
3. Seleção durante a passagem
Pesquisas básicas envolvendo a passagem de coronavírus tipo SARS-CoV de morcego em cultura de células e / ou modelos de animais estão em andamento há muitos anos em laboratórios de nível 2 de biossegurança em todo o mundo 27 , e existem casos documentados de fugas laboratoriais de SARS-CoV 28 . Portanto, devemos examinar a possibilidade de uma liberação laboratorial inadvertida do SARS-CoV-2.
Em teoria, é possível que o SARS-CoV-2 tenha adquirido mutações na RBD (Fig. 1a ) durante a adaptação à passagem na cultura de células, como foi observado em estudos sobre o SARS-CoV 11 . A descoberta de coronavírus do tipo SARS-CoV de pangolins com RBDs quase idênticos, no entanto, fornece uma explicação muito mais forte e parcimoniosa de como o SARS-CoV-2 os adquiriu por recombinação ou mutação 19 .
A aquisição do local de clivagem polibásica e dos glicanos ligados ao O previstos também argumenta contra cenários baseados em cultura. Novos locais de clivagem polibásica foram observados somente após a passagem prolongada do vírus da influenza aviária de baixa patogenicidade in vitro ou in vivo 17. Além disso, uma geração hipotética de SARS-CoV-2 por cultura celular ou passagem de animais exigiria o isolamento prévio de um vírus progenitor com similaridade genética muito alta, o que não foi descrito. A geração subsequente de um local de clivagem polibásica teria requerido passagem repetida em cultura de células ou animais com receptores ACE2 semelhantes aos de humanos, mas esse trabalho também não foi descrito anteriormente. Finalmente, é improvável que a geração dos glicanos O-preditos tenha ocorrido devido à passagem da cultura de células, pois essas características sugerem o envolvimento de um sistema imunológico 18 .
Conclusões
No meio da emergência global de saúde pública COVID-19, é razoável imaginar por que as origens da pandemia são importantes. A compreensão detalhada de como um vírus animal ultrapassou os limites das espécies para infectar seres humanos de maneira tão produtiva ajudará na prevenção de futuros eventos zoonóticos. Por exemplo, se o SARS-CoV-2 for pré-adaptado em outra espécie animal, haverá o risco de futuros eventos de reemergência. Por outro lado, se o processo adaptativo ocorreu em seres humanos, mesmo que ocorram transferências zoonóticas repetidas, é improvável que decolem sem a mesma série de mutações. Além disso, a identificação dos parentes virais mais próximos da SARS-CoV-2 circulando em animais ajudará bastante os estudos da função viral. De fato, a disponibilidade da sequência de morcegos RaTG13 ajudou a revelar as principais mutações RBD e o local de clivagem polibásica.
As características genômicas descritas aqui podem explicar em parte a infecciosidade e transmissibilidade da SARS-CoV-2 em humanos. Embora as evidências mostrem que o SARS-CoV-2 não é um vírus propositalmente manipulado, atualmente é impossível provar ou refutar as outras teorias de sua origem descritas aqui. No entanto, uma vez que observamos todos os recursos notáveis do SARS-CoV-2, incluindo o local otimizado de RBD e clivagem polibásica, nos coronavírus relacionados na natureza, não acreditamos que qualquer tipo de cenário laboratorial seja plausível.
Mais dados científicos podem balançar o balanço de evidências para favorecer uma hipótese sobre outra. Obter seqüências virais relacionadas de fontes animais seria a maneira mais definitiva de revelar as origens virais. Por exemplo, uma observação futura de um local de clivagem polibásico intermediário ou totalmente formado em um vírus do tipo SARS-CoV-2 de animais daria ainda mais suporte às hipóteses de seleção natural. Também seria útil obter mais dados genéticos e funcionais sobre SARS-CoV-2, incluindo estudos em animais. A identificação de um potencial hospedeiro intermediário de SARS-CoV-2, bem como o seqüenciamento do vírus em casos muito precoces, também seriam altamente informativos. Independentemente dos mecanismos exatos pelos quais o SARS-CoV-2 se originou por seleção natural,
Referências
- 1
- 2)
- 3)
- 4)
- 5)Dong, E., Du, H. & Gardner, L. Lancet Infect. Dis. https://doi.org/10.1016/S1473-3099(20)30120-1 (2020).
- 6Corman, VM, Muth, D., Niemeyer, D. & Drosten, C. Adv. Virus Res. 100 , 163-188 (2018).
- 7)Wan, Y., Shang, J., Graham, R., Baric, RS & Li, F.J. Virol. https://doi.org/10.1128/JVI.00127-20 (2020).
- 8)
- 9
- 10)Letko, M., Marzi, A. e Munster, V. Nat. Microbiol. https://doi.org/10.1038/s41564-020-0688-y (2020).
- 11)Sheahan, T. et ai. J. Virol. 82 , 2274- 2285 (2008).
- 12)Nao, N. et al. MBio 8 , e02298-16 (2017).
- 13)Chan, C.-M. et al. Exp. Biol. Med. 233 , 1527–1536 (2008).
- 14)Follis, KE, York, J. & Nunberg, JH Virology 350 , 358–369 (2006).
- 15
- 16Alexander, DJ e Brown, IH Rev. Sci. Tech. 28 , 19-38 (2009).
- 17Ito, T. et al. J. Virol. 75 , 4439- 4443 (2001).
- 18Bagdonaite, I. & Wandall, HH Glycobiology 28 , 443–467 (2018).
- 19Cui, J., Li, F. & Shi, Z.-L. Nat. Rev. Microbiol. 17 , 181-192 (2019).
- 20Almazán, F. et al. Virus Res. 189 , 262-270 (2014).
- 21
- 22)Yamada, Y. & Liu, DX J. Virol. 83 , 8744-8758 (2009).
- 23
- 24)
- 25)Dudas, G., Carvalho, LM, Rambaut, A. & Bedford, T. eLife 7 , e31257 (2018).
- 26)Wang, N. et ai. Virol. Pecado. 33 , 104-107 (2018).
- 27Ge, X.-Y. et al. Nature 503 , 535-538 (2013).
- 28)Lim, PL et al. N. Engl. J. Med. 350 , 1740-1745 (2004).
- 29Wong, MC, Javornik Cregeen, SJ, Ajami, NJ & Petrosino, JF bioRxiv https://doi.org/10.1101/2020.02.07.939207 (2020).
- 30)Liu, P., Chen, W. & Chen, J.‑P. Vírus 11 , 979 (2019).
Reconhecimentos
Agradecemos a todos aqueles que contribuíram com seqüências para o banco de dados GISAID ( https://www.gisaid.org/ ) e as análises para Virological.org ( http://virological.org/ ). Agradecemos a M. Farzan pelas discussões e ao Wellcome Trust pelo apoio. O KGA é um Pew Biomedical Scholar e é apoiado pelo NIH grant U19AI135995. O AR é apoiado pelo Wellcome Trust (rede de colaboradores 206298 / Z / 17 / Z - ARTIC) e pelo Conselho Europeu de Pesquisa (contrato de concessão nº 725422 - ReservoirDOCS). A ECH é apoiada por uma bolsa de estudos da ARC Australian Laureate (FL170100022). O RFG é suportado pelas bolsas do NIH U19AI135995, U54 HG007480 e U19AI142790.
Informação sobre o autor
Declarações de ética
Interesses competitivos
A RFG é cofundadora da Zalgen Labs, uma empresa de biotecnologia que desenvolve contramedidas para vírus emergentes.
Direitos e permissões
Sobre este artigo

Citar este artigo
Andersen, KG, Rambaut, A., Lipkin, WI et al. A origem proximal do SARS-CoV-2. Nat Med (2020). https://doi.org/10.1038/s41591-020-0820-9
- Publicados
Sem comentários:
Enviar um comentário